单基因TCGA的Cox森林图怎么画才好看呢
转自我的简书https://www.jianshu.com/p/145b2dc338a0
首先调取Xena网页的TCGA数据做Cox生存分析,数据可以通过hiplot官网自主研发的ucsc-xena-shiny直接在线获取
访问https://hiplot.com.cn/advance/ucsc-xena-shiny
然后全屏显示,点击Qucik PanCan Analysis下方的TCGA:Molcular Profile Cox Analysis
输入一个你想要的基因,比如RAC3,Select Measure for plot可以设置OS,PFI,DSS和DFI,然后点上方的搜索🔍,就可以看到出的图了
继续往下滚动鼠标,就可以看到数据了,而且还可以下载
得到数据以后就可以用R画图了,注意,这里的HR和CI都是Log过的结果,跟别的地方计算的Cox结果有些不一样,可能是方法不一样吧,是因为网站计算的HR结果相差太大了吗?
由于是log过的结果,所以森林图的X轴不再是HR=1为分界线了,而是以log2HR=0为分界线。。。
RAC3_mRNA_OS_pancan_unicox <- read.csv("~/Desktop/RAC3_mRNA_OS_pancan_unicox.csv") ##加载csv数据
library(ggplot2)
ggplot(RAC3_mRNA_OS_pancan_unicox, aes(HR_log, cancer, col=Type))+ ##定义X轴和Y轴,以类型分类
geom_point(size=2.5)+ #固定点的大小
geom_errorbarh(aes(xmax =upper_95_log, xmin = lower_95_log), height = 0.4)+ ##设置95%CI区间,就是误差线
scale_x_continuous(limits= c(-2, 2), breaks= seq(-1, 1, 1))+ ##设置X轴范围,分割点从-1到1,以1为分界,具体分界看数字分布
geom_vline(aes(xintercept = 0))+ #以0为分界线
xlab('HR(95%CI)') + ylab(' ')+ #定义标签
theme_bw(base_size = 12)+ #主题和字体
scale_color_manual(values = c("gray", "steelblue", "brown")) #设置颜色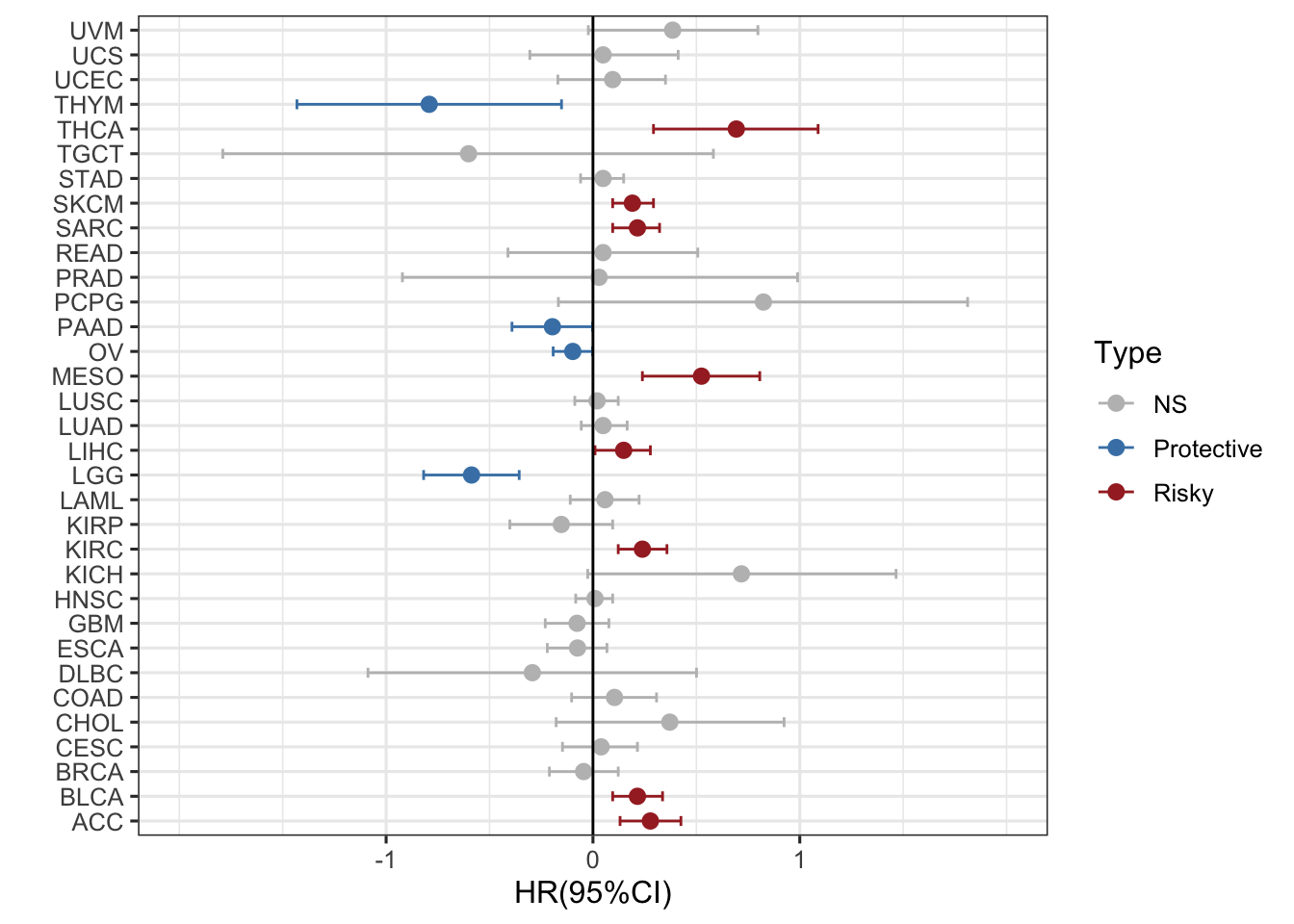 #### 可以换一下不同的形状表示不同的状态,用的是
#### 可以换一下不同的形状表示不同的状态,用的是shape=Type
ggplot(RAC3_mRNA_OS_pancan_unicox, aes(HR_log, cancer, col=Type,shape=Type))+ #设置不同的形状
geom_point(size=3)+
geom_errorbarh(aes(xmax =upper_95_log, xmin = lower_95_log), height = 0.4)+
scale_x_continuous(limits= c(-2, 2), breaks= seq(-1, 1, 1))+
geom_vline(aes(xintercept = 0))+
xlab('HR(95%CI)') + ylab(' ')+
theme_bw(base_size = 12)+
scale_color_manual(values = c("gray", "steelblue", "brown"))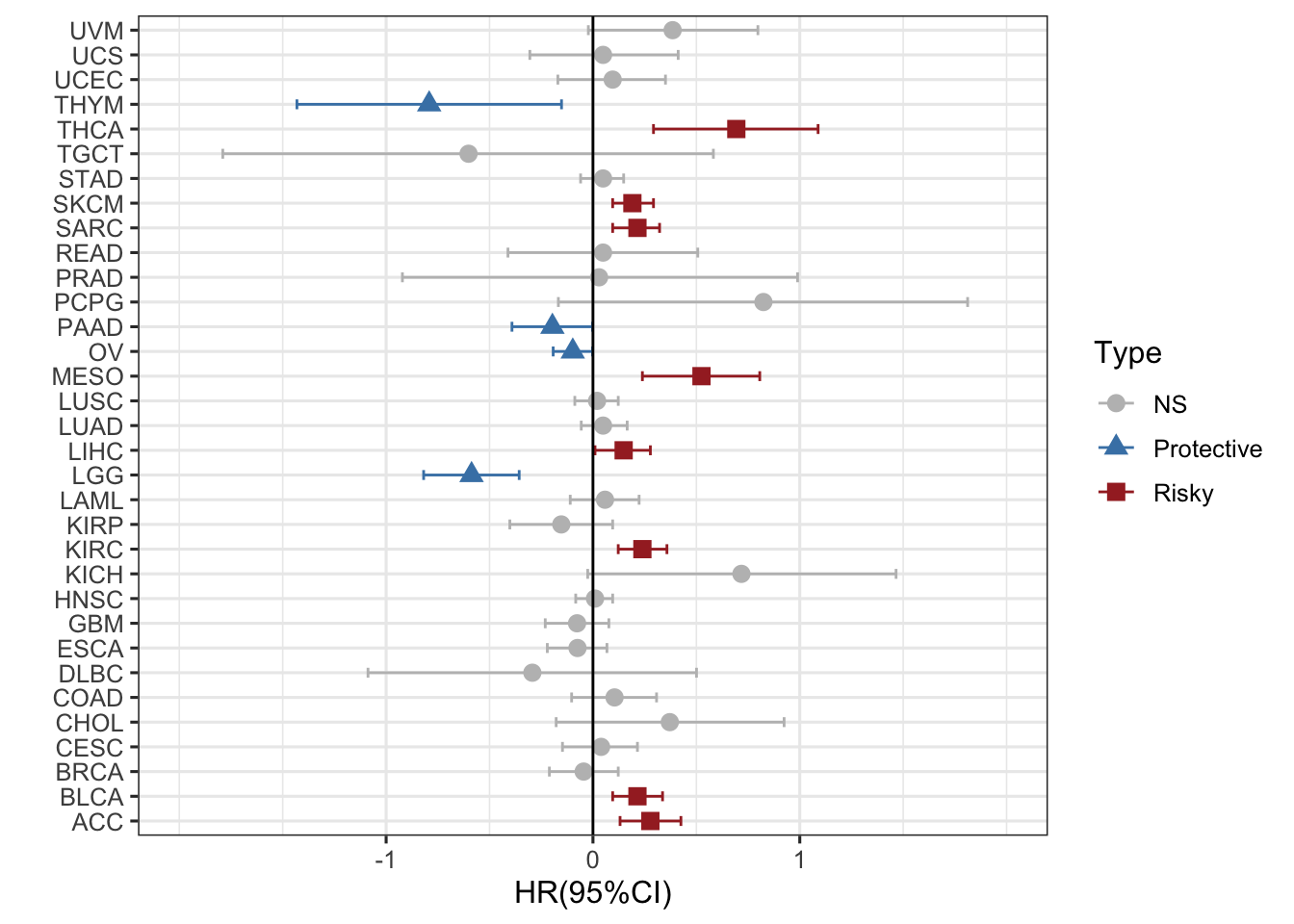 ### 这个图看起来所有的形状和点都一样大,用什么办法区别P值,类似火山图,我们可以取-log10P值,这样越大就越显著
### 这个图看起来所有的形状和点都一样大,用什么办法区别P值,类似火山图,我们可以取-log10P值,这样越大就越显著
# 以-log10P值定义点的大小,点越大,P值越小,越有统计学意义
ggplot(RAC3_mRNA_OS_pancan_unicox, aes(HR_log, cancer, col=Type))+
geom_point(aes(size=-log10(p.value)))+
geom_errorbarh(aes(xmax =upper_95_log, xmin = lower_95_log), height = 0.4)+
scale_x_continuous(limits= c(-2, 2), breaks= seq(-1, 1, 1))+
geom_vline(aes(xintercept = 0))+
xlab('HR(95%CI)') + ylab(' ')+
theme_bw(base_size = 12)+
scale_color_manual(values = c("gray", "steelblue", "brown"))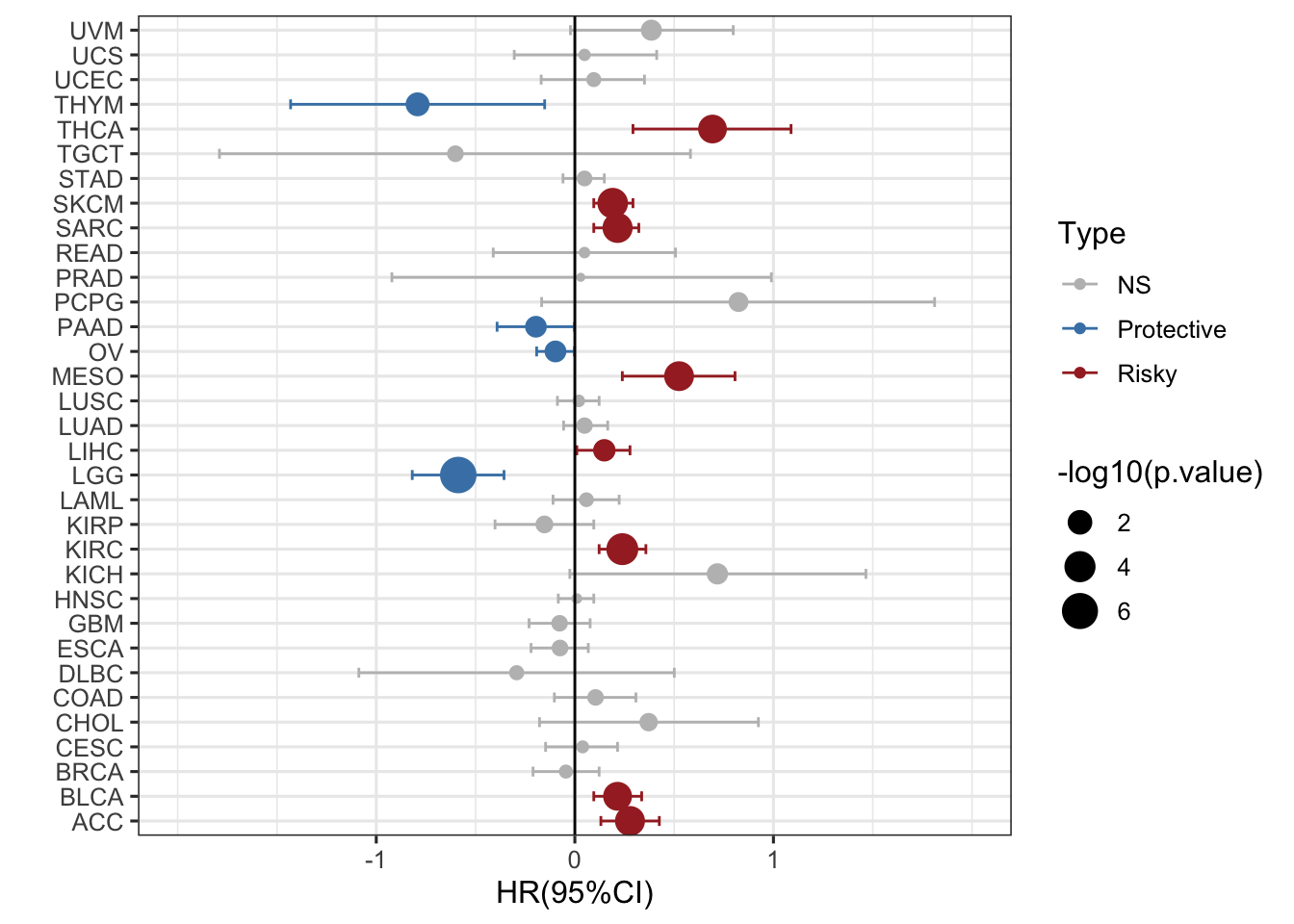 ### 再加一个形状
### 再加一个形状
ggplot(RAC3_mRNA_OS_pancan_unicox, aes(HR_log, cancer, col=Type,shape=Type))+
geom_point(aes(size=-log10(p.value)))+
geom_errorbarh(aes(xmax =upper_95_log, xmin = lower_95_log), height = 0.4)+
scale_x_continuous(limits= c(-2, 2), breaks= seq(-1, 1, 1))+
geom_vline(aes(xintercept = 0))+
xlab('HR(95%CI)') + ylab(' ')+
theme_bw(base_size = 12)+
scale_color_manual(values = c("gray", "steelblue", "brown"))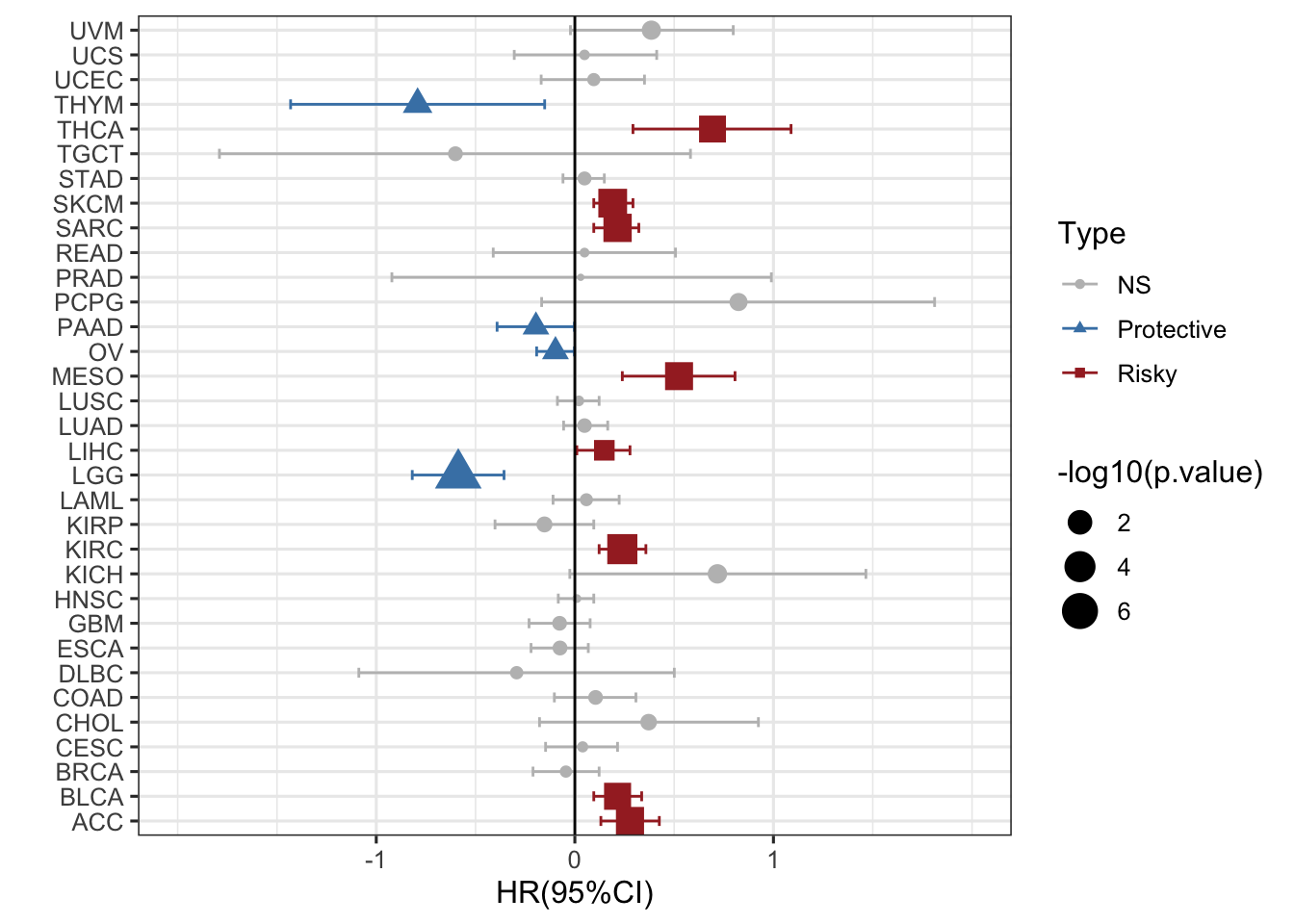
看起来很凌乱,排个序可好,用的是reorder函数
ggplot(RAC3_mRNA_OS_pancan_unicox, aes(HR_log, reorder(cancer,HR_log), col=Type,shape=Type))+
geom_point(aes(size=-log10(p.value)))+
geom_errorbarh(aes(xmax =upper_95_log, xmin = lower_95_log), height = 0.4)+
scale_x_continuous(limits= c(-2, 2), breaks= seq(-1, 1, 1))+
geom_vline(aes(xintercept = 0))+
xlab('HR(95%CI)') + ylab(' ')+
theme_bw(base_size = 12)+
scale_color_manual(values = c("gray", "steelblue", "brown"))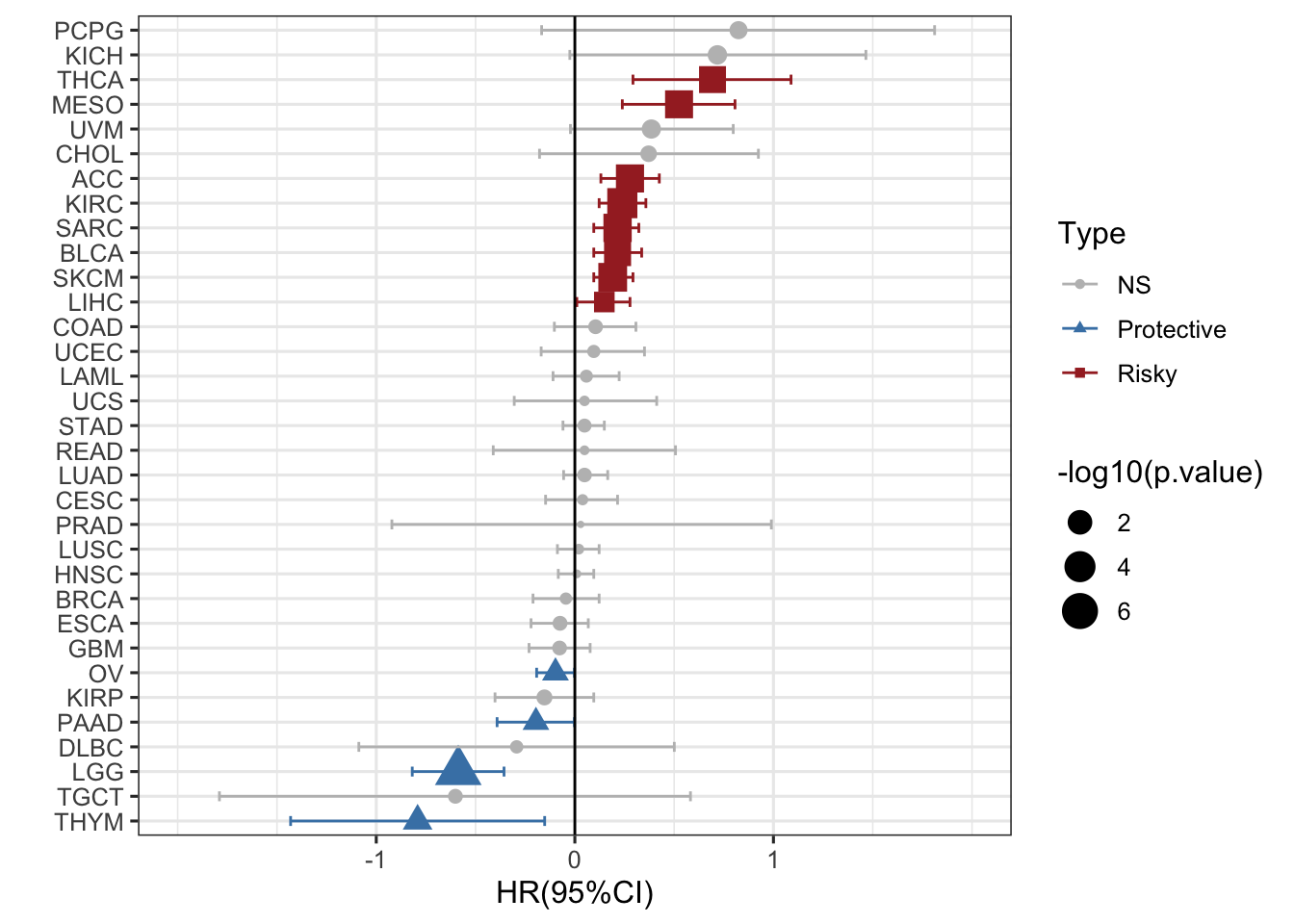
换个排序方式也行
ggplot(RAC3_mRNA_OS_pancan_unicox, aes(HR_log, reorder(cancer,-HR_log), col=Type,shape=Type))+
geom_point(aes(size=-log10(p.value)))+
geom_errorbarh(aes(xmax =upper_95_log, xmin = lower_95_log), height = 0.4)+
scale_x_continuous(limits= c(-2, 2), breaks= seq(-1, 1, 1))+
geom_vline(aes(xintercept = 0))+
xlab('HR(95%CI)') + ylab(' ')+
theme_bw(base_size = 12)+
scale_color_manual(values = c("gray", "steelblue", "brown"))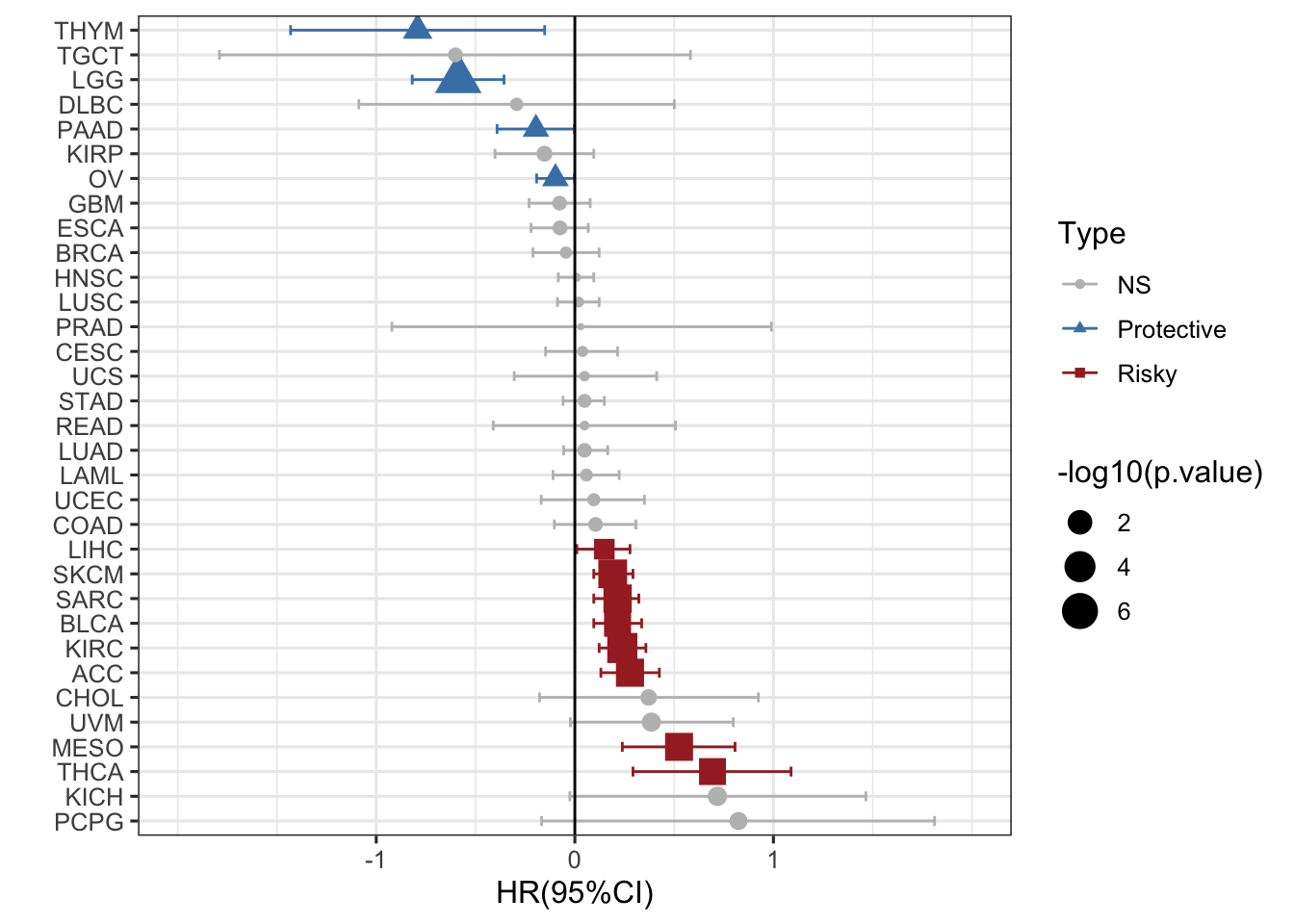
更多定制,等你发现。。。
欧阳松
主治医师、讲师
My research interests include urogenital tumors, urolithiasis, male infertility, male erectile dysfunction,etc.